Paper: DrugCLIP: Contrastive Protein-Molecule Representation Learning for Virtual Screening
In the ever-evolving landscape of drug discovery, the quest for efficiency and precision in Virtual screening (VS) has led to the development of DrugCLIP.
Comparing to Traditional Methods
- Docking simulations: Restricted library, highly time-consuming (~ 3s/molecule). It is impractical when dealing with large libraries ($10^9$).
- Supervised learning methods: Strong dependency on limited data with reliable binding-affinity labels. Also restricted by the scarcity of reliable negative samples. Have not yet surpassed docking methods.
- DrugCLIP: A dense retrieval approach (inspired by CLIP), uses contrastive learning to align representations of binding protein pockets and molecules without explicit binding-affinity scores. DrugCLIP allows for offline pre-computation of protein and molecule encodings, bringing high efficiency to online inference on billions of molecules.
How Does DrugCLIP Work?
Problem Setting
Old virtual screening paradigm: Given a protein pocket $p$ and a set of small molecules $M={m_1, m_2, \dots, m_n}$, the objective of virtual screening is to identify the top $k$ candidates with the highest probability of binding to the target pocket, guided by a scoring function that assesses the pairwise data between $p$ and $m_i$.
Our work views it as a dense retrieval task. Reformulating virtual screening as a similarity matching problem: Given a protein pocket as the query, we aim to retrieve from a large-scale molecule library the most relevant molecules with the highest probability of binding to the target pocket.
Framework
At the heart of DrugCLIP lies the CLIP (Contrastive Language–Image Pre-training) framework. Originally designed for understanding and correlating textual and visual information, CLIP’s principles have been adapted to serve the unique context of drug discovery. It learns to distinguish between matching and non-matching pairs of protein and molecule representations.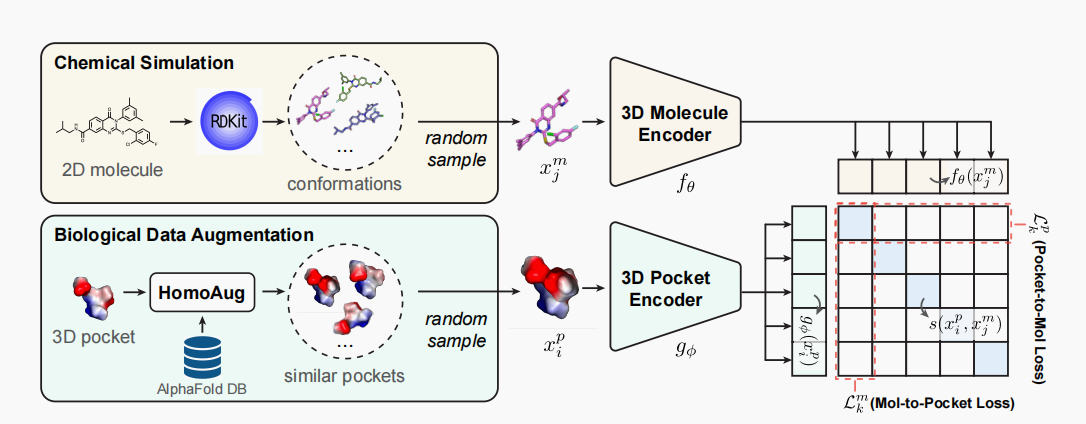
Encoders
- The architecture is followed by UniMol.
Objective
- Similarity score: Dot product of the representation of protein $x^p$ and molecule $x^m$.
- In-batch sampling strategy for negative and positive pairs.
Pocket-to-Mol loss function:
$$
L_{pk}(x_{pk}, {x_{mi}}^N_{i=1}) = -\frac{1}{N} \log \frac{\exp(s(x_{pk}, x_{mk})/\tau)}{\sum^N_{i=1} \exp(s(x_{pk}, x_{mi})/\tau)}
$$
- $L_{pk}$: Pocket-to-Mol loss for a given protein $x_{pk}$.
- $x_{pk}$: The protein of interest in the pair.
- $x_{mk}$: The correct binding molecule for protein $x_{pk}$.
- $x_{mi}$: Molecules in the dataset, where $i$ ranges from 1 to $N$, including the correct binding molecule $x_{mk}$.
- $s(x_{pk}, x_{mi})$: Similarity score between protein $x_{pk}$ and molecule $x_{mi}$, calculated using a similarity function such as dot product or cosine similarity.
- $\tau$: Temperature parameter in the softmax function, adjusting the distribution’s sharpness.
- $N$: Batch size, representing the number of protein-molecule pairs in the dataset.
Mol-to-Pocket loss:
$$L_{mk}(x_{mk}, {x_{pi}}^N_{i=1}) = -\frac{1}{N} \log \frac{\exp(s(x_{pk}, x_{mk})/\tau)}{\sum^N_{i=1} \exp(s(x_{pi}, x_{mk})/\tau)}$$
Final Loss:
$$ L = \frac{1}{2} \sum_{k=1}^{N} (L_{pk} + L_{mk}) $$
Data
Trainning Data
One of the advantages of our method is that we can facilitate the usage of large-scale unlabeled data beyond densely annotated small datasets such as PDBBind, BioLip, and ChEMBL.
Dataset Complexes Preprocess Link PDBBind 2019 >17000 protein-ligand complex structures along with their binding-affinity labels PDBBind 2019 BioLip 122861 filter out all complexes that contain peptides, DNA, RNA, and single ions BioLip ChEMBL around 1000 use proteins with only one known binding pocket, pair the pocket with all positive binders ChEMBL We also introduce a biological-knowledge inspired augmentation method, HomoAug.
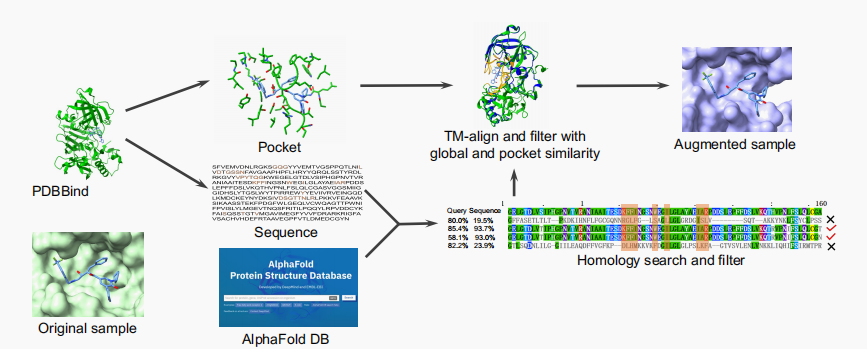
The pocket protein instances from PDBBind are searched for homologous counterparts in the AlphaFold Protein Structure Database. Then, the TMalign method is employed to achieve structural alignment between the homologous protein and the original protein.
Experiments
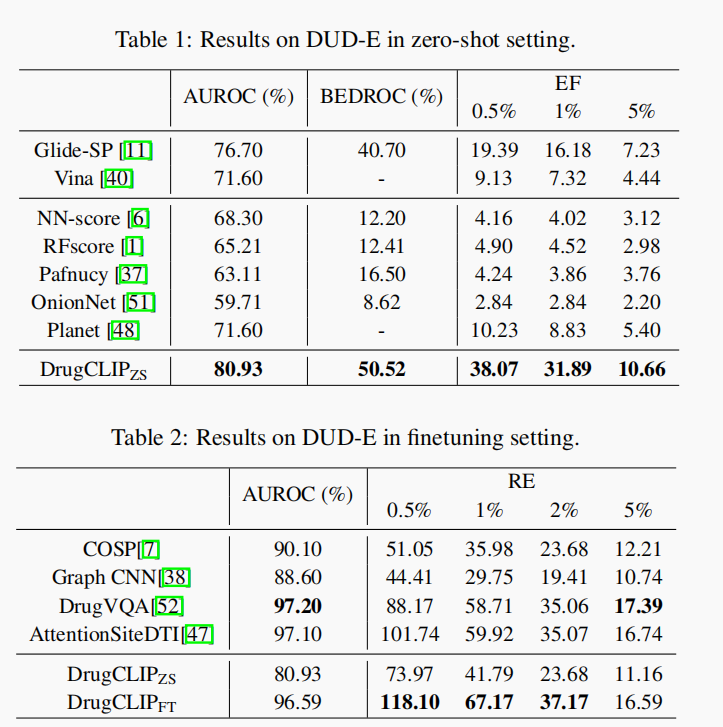
- Evaluation on DUD-E Benchmark
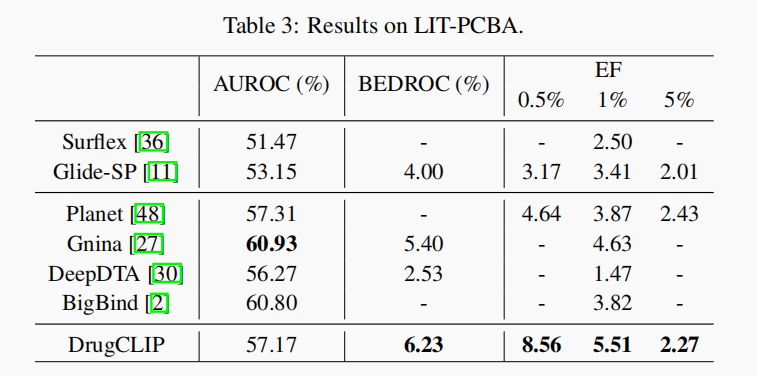
- Evaluation on LIT-PCBA Benchmark
- Efficiency Analysis for Large-scale Virtual Screening
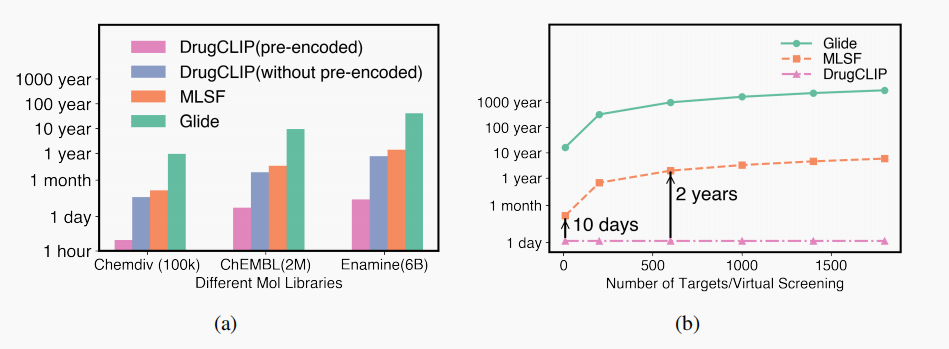
The Future of DrugCLIP
As we continue to refine and expand the capabilities of DrugCLIP, its potential to accelerate the drug discovery process is immense. By offering a more reliable, efficient, and scalable solution, DrugCLIP paves the way for the identification of novel drug candidates.