Overview
Tandem mass spectrometry (MS/MS) is the cornerstone of modern proteomics, enabling large-scale protein identification through peptide-spectrum matching. Open search strategies, which allow for arbitrary post-translational modifications (PTMs) by searching with wide precursor mass tolerance windows (±500 Da), face severe computational challenges due to the combinatorial explosion of candidate peptides.
We present a novel biology-informed hybrid search engine that combines adaptive approximate nearest neighbor (ANN) search with domain-specific optimizations for high-dimensional vector similarity search under mass range constraints.
The Open Search Challenge
Traditional database search strategies assume that peptides exist in their canonical, unmodified forms. However, post-translational modifications (PTMs) such as phosphorylation, acetylation, and oxidation are ubiquitous in biological systems and alter peptide masses. Open search addresses this by allowing large precursor mass tolerance windows (typically ±500 Da), enabling the discovery of unknown or unexpected modifications without prior specification.
This generality comes at a steep computational cost. The number of candidates explodes as the mass tolerance window $\Delta$ increases, creating a severe computational bottleneck when combined with high-dimensional vector similarities computation.
We formulate the peptide identification problem as a range-filtering approximate nearest neighbor search (RF-ANNS):
$$ \mathcal{R}(q, \Delta, k) = \underset{p \in \mathcal{F}(q, \Delta)}{\arg\max_k} \text{sim}(\mathbf{v}_q, \mathbf{v}_p) $$
where:
- $\mathcal{F}(q, \Delta) = {p \in \mathcal{D} : |m_p - m_q| \leq \Delta}$ is the mass filter
- $\text{sim}(\cdot, \cdot)$ measures cosine similarity between spectrum and peptide($p$) embeddings
- $k$ is the number of top candidates to retrieve
This formulation reveals a fundamental tension: low-selectivity queries require efficient filtering, while high-selectivity queries demand fast similarity computation.
Contributions
Our system introduces three key innovations:
- Biology-Informed Data Layout: A columnar storage format sorted by precursor mass that reduces range query complexity from $O(|\mathcal{D}|)$ hash lookups to $O(\log |\mathcal{D}| + |\mathcal{F}|)$ via binary search, with superior I/O efficiency.
- Rescue-HNSW Algorithm: An adaptive ANN search strategy that maintains graph connectivity in pre-filtered scenarios by injecting mass-proximate neighbors, addressing the “connectivity crisis” in filtered graph traversal.
- Selectivity-Adaptive Execution: A dynamic query optimizer that switches between post-filtering HNSW, and pre-filtering Rescue-HNSW based on estimated selectivity.
Methodology
System Architecture
Our system comprises three tightly integrated layers designed for selectivity-adaptive query processing:
- Storage Layer: Columnar database with peptides sorted by precursor mass, enabling $O(\log N)$ binary search for range filtering. Embeddings are stored in memory-mapped arrays for sequential access.
- Index Layer: HNSW graph augmented with mass-aware neighbor lists. Each node maintains both semantic neighbors and mass neighbors, ensuring connectivity at low selectivity.
- Query Engine: Adaptive executor that routes queries to one of three strategies based on estimated selectivity.
Biology-Informed Hybrid Graph & Rescue-HNSW
Pre-filtering creates a subgraph induced by valid mass nodes. However, this subgraph may be disconnected when selectivity is low, causing graph search to get trapped in local neighborhoods.
Our key insight: mass proximity implies potential semantic relevance in proteomics. To preserve connectivity while enabling efficient filtering, we augment each node with a second neighbor list:
- Semantic neighbors ($M = 32$): Standard HNSW edges to the closest nodes in embedding space.
- Mass neighbors ($M’ = 32$): Edges to nodes that are within a narrow mass window ($\pm 20$ ppm) and close in embedding space.
During the Rescue-HNSW algorithm, we dynamically inject these mass neighbors to maintain network connectivity:
Adaptive Query Execution
Our query optimizer estimates selectivity $\hat{\sigma}$ exactly at query time via binary search, and routes queries to one of three strategies based on empirically-determined thresholds:
- Brute-Force GPU or Hybrid Graph Traversal (Rescue-HNSW) ($\hat{\sigma} < \tau_1$): Handle extremely small candidate sets.
- Hybrid Graph Traversal ($\tau_1 \leq \hat{\sigma} \leq \tau_2$): Apply pre-filtering with mass-aware rescue (Rescue-HNSW).
- Adaptive Post-Filtering ($\hat{\sigma} > \tau_2$): Search full index with adaptive $k$-doubling.
Experimental Results
On a standard proteomics dataset (HEK293, 37,131 spectra against 4.87M peptides), our approach demonstrates significant improvements:
Query Throughput
Our system achieves up to 13× speedup over state-of-the-art tools like MSFragger. We maintain ~2,000 queries per second (QPS) even with extreme 500 Da tolerance windows.
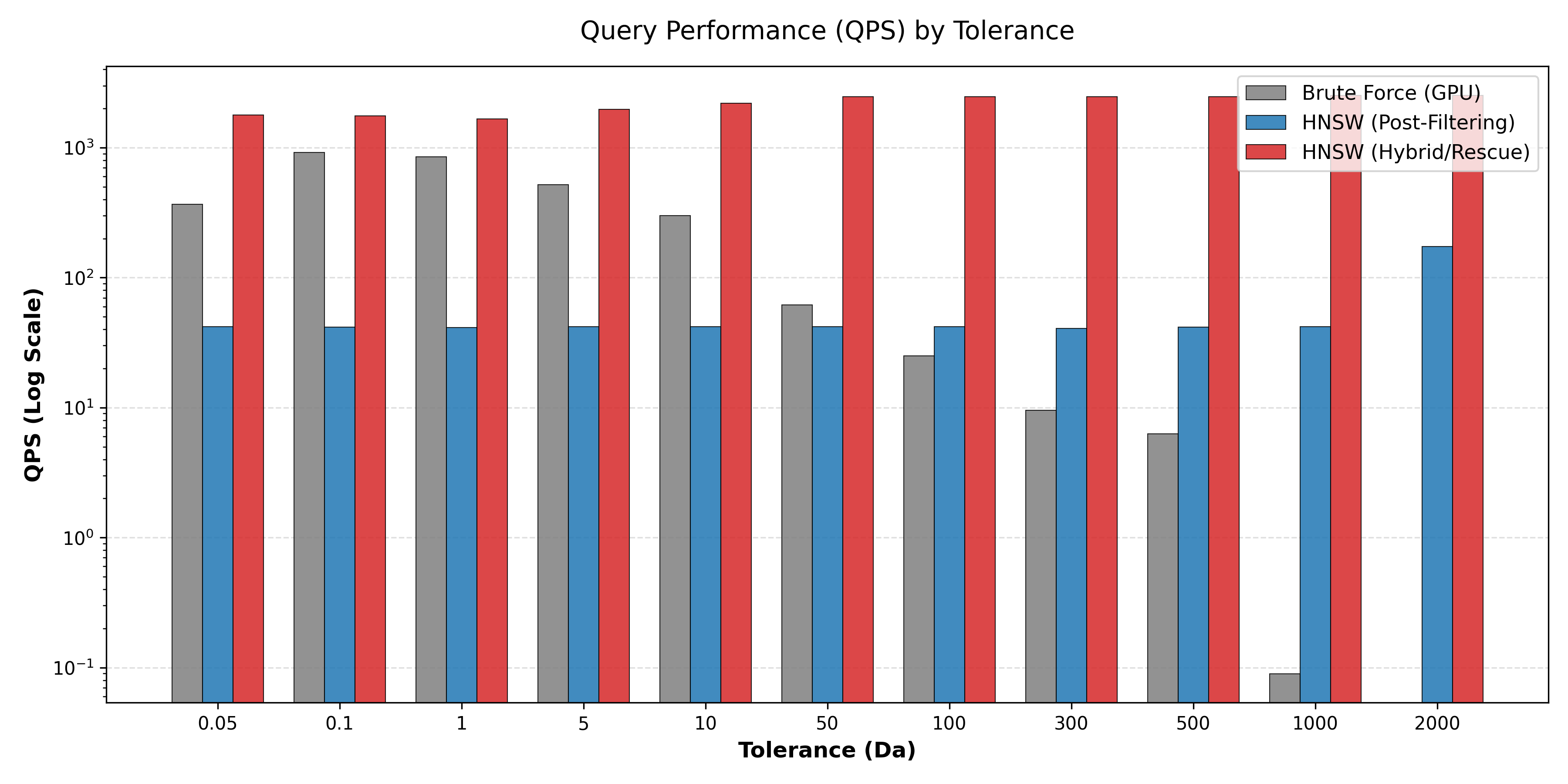
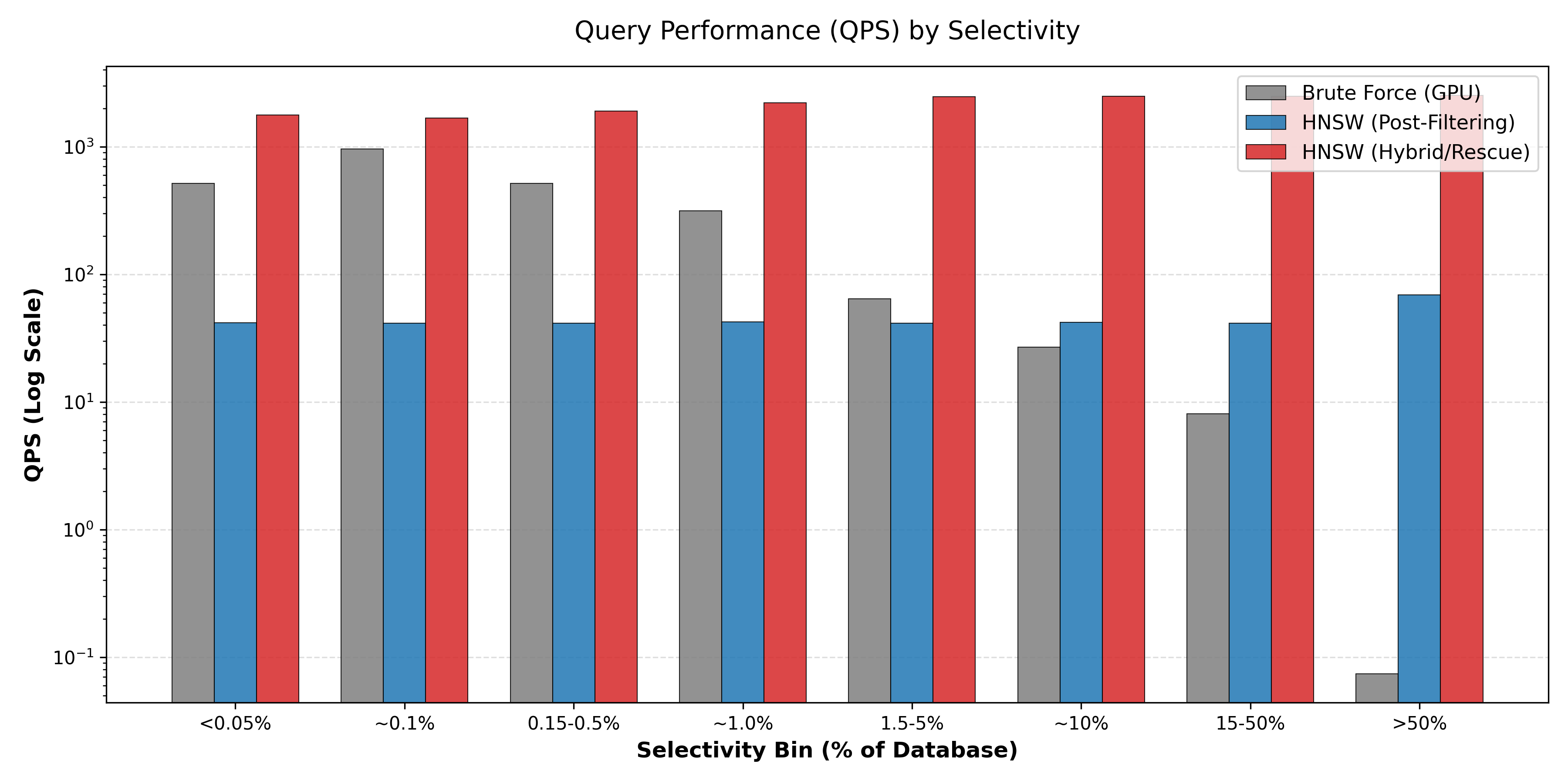
At large mass windows (1-100 Da and beyond), HNSW-v2 (Rescue-HNSW) clearly dominates the performance landscape by effectively balancing pre-filtering benefits against the cost of maintaining graph connectivity.
Identification Rate
We maintain a competitive identification rate under strict 1% FDR control.
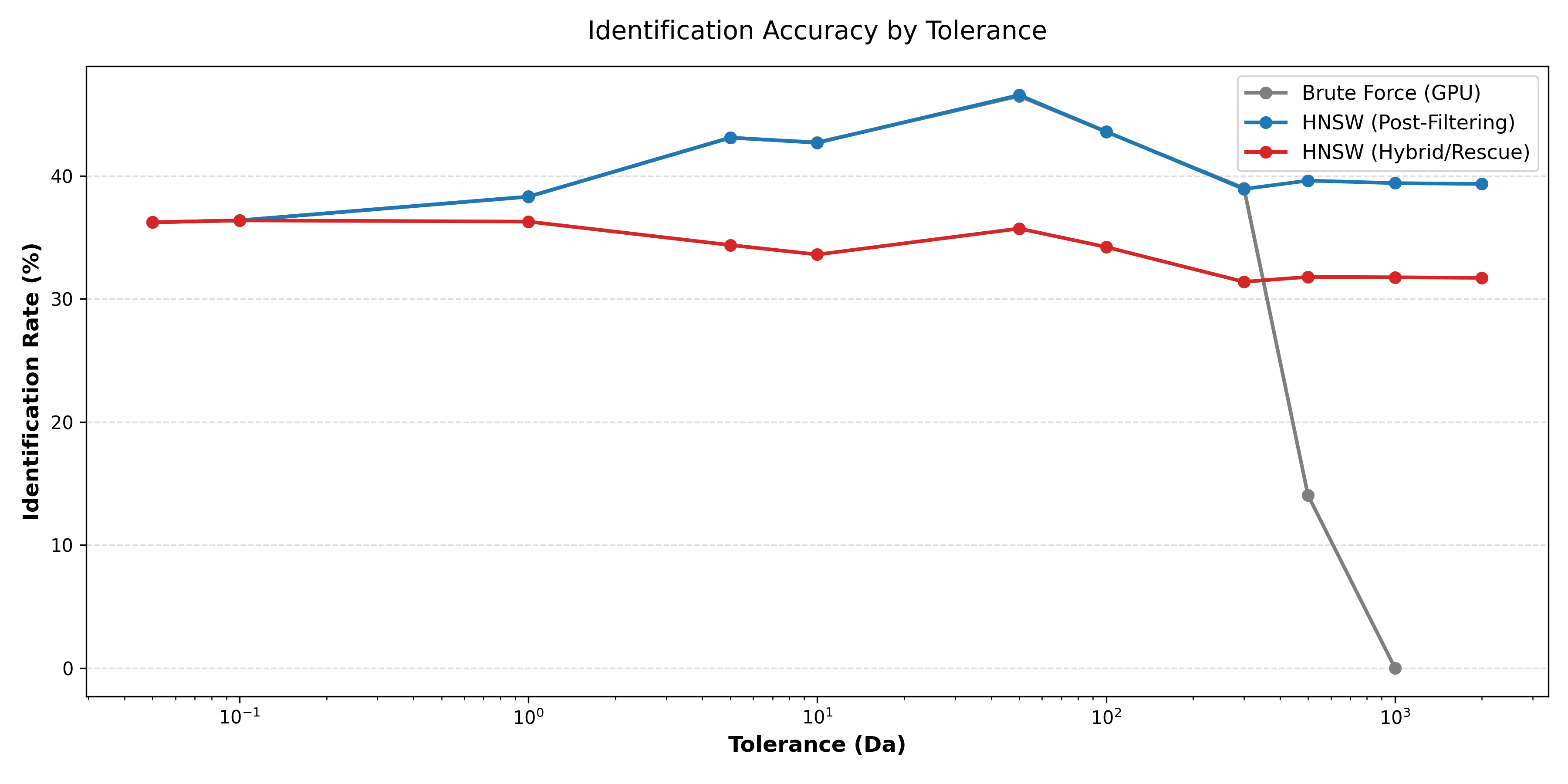
While post-filtering achieves higher recall due to a complete graph, HNSW-v2 offers a compelling balance of identification rate and extreme speed, making interactive open-search workflows viable.
Conclusion
By exploiting the physical constraint of precursor mass through columnar storage, and introducing Rescue-HNSW to preserve graph connectivity under aggressive filtering, we transform open search from a time-intensive batch operation into an interactive analytical tool. Our system demonstrates exactly how principled integration of biological constraints with modern ANN algorithms can accelerate real-world scientific discovery.